-
Viewpoint on 'IONS'
Viewpoint on 'Scientific Literacy'
- Proudly sponsored by
-

-
Listen to the Light and See Deeper

Imaging fluorescent proteins in thick biological samples has always been a challenge. Now a new technique has harnessed the opto-acoustic effect in order to make deep tissue imaging easier for biologists.
-
Magnetic Break Up
We usually think of the north and south magnetic poles as an inseparable couple. Experiments have now shown that under the appropriate conditions they can, in fact, break up.
-
Traffic Lights for Chemotherapy

Many cancer patients do not respond to chemotherapy and have to endure harmful side effects even when the treatment itself does not prove effective. A new technique could, as of the very first day, give the green or red light to continuing with the treatment.
Volume 4 Story 1 - 21/1/2009
Many Photons get More
Nanoscopy, optical microscopy with 10-30 nm detail, has become a reality; in fact it has been chosen “Method of the year 2008” by Nature Methods [1]. We are all aware of the wave nature of light and the ensuing diffraction limit for resolution. So have the rules of physics been broken? What’s the trick and where’s the catch?
 This year it is exactly 400 years since Galileo first used a telescope. With his few-cm lens Galileo gained sufficient resolution to observe the moons of Jupiter, the phases of Venus and the handles of Saturn. In astronomy, size makes the difference: the angular resolution scales with the number of wavelengths covered by the collecting objective. Modern giant telescopes with their 10 m mirrors span over 107 optical wavelengths, thus providing diffraction-limited resolution of 10-7 radians (~0.01 arcseconds) and enormous light collection power, sufficient to distinguish exoplanets and the most red-shifted galaxies. Looking at the microscopic world, again the wave nature of light sets the limit. To resolve details it is important to capture and refocus the full angular spectrum of the light of an object, i.e. all spatial frequencies up to the maximum wavevector 2π/λ. For larger wavevectors the wave is evanescent, eventually lost and not captured by the objective. The smallest feature Δx that can be distinguished has a direct Fourier connection to the highest frequency: Δx ≥ λ/2. Hence, for conventional lens-based high resolution optical microscopy the resolution is typically 200-300 nm. Modern super-resolution optical microscopy goes far beyond that, revealing 10-30 nm details. In this context one comes across several new ingredients: single molecule detection, photo-activation, photo-switching, state depletion, super-oscillations, plasmonic focussing, near field optics and super-lensing. What is the new concept?
This year it is exactly 400 years since Galileo first used a telescope. With his few-cm lens Galileo gained sufficient resolution to observe the moons of Jupiter, the phases of Venus and the handles of Saturn. In astronomy, size makes the difference: the angular resolution scales with the number of wavelengths covered by the collecting objective. Modern giant telescopes with their 10 m mirrors span over 107 optical wavelengths, thus providing diffraction-limited resolution of 10-7 radians (~0.01 arcseconds) and enormous light collection power, sufficient to distinguish exoplanets and the most red-shifted galaxies. Looking at the microscopic world, again the wave nature of light sets the limit. To resolve details it is important to capture and refocus the full angular spectrum of the light of an object, i.e. all spatial frequencies up to the maximum wavevector 2π/λ. For larger wavevectors the wave is evanescent, eventually lost and not captured by the objective. The smallest feature Δx that can be distinguished has a direct Fourier connection to the highest frequency: Δx ≥ λ/2. Hence, for conventional lens-based high resolution optical microscopy the resolution is typically 200-300 nm. Modern super-resolution optical microscopy goes far beyond that, revealing 10-30 nm details. In this context one comes across several new ingredients: single molecule detection, photo-activation, photo-switching, state depletion, super-oscillations, plasmonic focussing, near field optics and super-lensing. What is the new concept?
It is the capacity to detect and image single molecules at room temperature that has triggered ideas towards molecular optical imaging. Essentially, the position of a single isolated molecule can be determined with very high accuracy, limited only by the amount of detected signal. In fact, if N photons are detected, Poison statistics tells us that the location precision improves with N1/2, (assuming other noise terms can be ignored). For example, for 1000 counted photons the position accuracy is ~3% of the actual spatial response, which comes down to 15 nm for a diffraction limited spot of 500 nm. Already in 1994, Eric Betzig [2] had stated that if each molecule could be discriminated by some unique optical feature, a map of their positions could be reconstructed and molecular resolution imaging would become possible. It took another decade before a natural discriminator for molecules was exploited: time. Single molecules do blink and bleach; a stochastic process where only a fraction of the molecules is on at a time. To get control over the irregular time behaviour, Betzig and colleagues introduced photoactivation: with a delicate optical stimulus they activated a tiny fraction of molecules, imaged this low density fraction of molecules until they all photo-bleached and then they determined their exact positions. This way a flag is set for each molecule that dares to show up. By successive iteration of this activation-mapping-bleach trick they built a position map. The map gets filled with flags, all at nanometric accuracy through single molecule localization: Photo-Activation Localization Microscopy (PALM) [2]. An alternative approach is to switch molecules on temporarily, without bleaching, a method known as Stochastic Optical Reconstruction Microscopy (STORM) [3]. In either case, photo-switching microscopy is finding rapid spreading of applications in a variety of biological domains. PALM and STORM are advertised as super-resolution techniques, revolutionizing microscopy by breaking the diffraction limit for microscopy. However, isnt this revolution a deconvolution? No, it is not a simple deconvolution with the microscopic spatial response function, as the time discrimination of single photon emitters is essential: truly photoswitching microscopy. Has the diffraction limit been broken? Definitely not; nevertheless one should clearly differentiate between diffraction limited microscopic resolution and photon yield limited localization accuracy. It is the combination of high resolution and high signal that yields super localisation: all according to the rules of diffraction and photon statistics. Thus, the result of localization microscopy is a detailed map of discrete molecular positions, a reconstructed image, built-up through a stochastic process. The catch is that one relies on photoswitchable fluorescent molecules, detected at the level of single molecules, which are by necessity bleached for localization mapping.
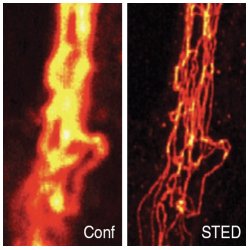
The advantage of photoswitching in microscopy is clear, but can it also be made to work effectively at the ensemble level beyond single molecule detection? The loss of single molecule localization certainly requires a radically different approach. As far back as 1994, Stefan Hell [4] had already demonstrated that a fluorescent sample can be switched off by depleting the excited state through stimulated emission. Interestingly, through the molecular photodynamics, the reduction of fluorescence shows a highly non-linear response on the depletion power. This non-linearity now is the key to recovering spatial accuracy: Stefan Hell and colleagues devised an ingenious two-color scheme with different spatial profiles for the excitation and depletion beams, leaving a non-depleted spot at the centre of the excitation profile. The trick is that the non-linearity squeezes the effective non-depleted fluorescent spot to a tiny area, down to ~15 nm. This has led to the success story of Stimulated Emission Depletion (STED) microscopy [4], now super-resolution method of the year. Is the diffraction limit broken? Of course both the excitation and the depletion beams perfectly obey the laws of diffraction. It is the non-linear STED response of the molecule that squeezes the effective spatial imaging response to far below the diffraction limit. Again, the role of the molecule is essential! More importantly, a real direct image is obtained with true super-resolution, which should not be confused with the localization reconstruction map obtained by PALM/STORM. For that reason, photon statistics is not a direct issue for the super-resolution in STED. Yet the catch is that photon yield stays part of the game, as not all fluorophores survive the intense depletion treatment and one needs to focus on relatively photostable molecules that survive the excitation-depletion photo-cycle.
Still, there is a soft edge to the diffraction limit. Recently, Michael Berry [5] showed that sub-wavelength optical fields can actually be created in the far field, at distances of many wavelengths. These so-called super-oscillations are a subtle phenomenon associated with delicate correlations between Fourier components. In principle, sub-wavelength focal points can be created at longer distances (not to be confused with the Talbot effect) without evanescent waves, based on these super-oscillations, containing spatial frequencies beyond the free space wave vector, i.e. faster than Fourier. Unfortunately the super-oscillations form a rapidly decaying tail of the total spatial frequency spectrum. Thus the high resolution components are exponentially weak in the super-oscillatory region and are surrounded by strong, low frequency components. If it wasnt for very good signal/noise allowing a high dynamic, one could not hope to discern some of the super-resolution aspects.
To break the diffraction limit, free of photodynamic properties of the sample, one needs to interact predominantly with the non-propagating, high spatial frequency contribution and by necessity move into the near field. Here, ironically, the sample becomes part of the microscope: one measures the spatial response of the mutual interaction and any super-resolution response cannot be specified as resolution of the microscope. For example, scanning a metallic nanoparticle around a luminescent quantum dot gives very informative nanoscale maps, but where is the microscope? A predominant challenge that remains in near field imaging is to confine the optical field to the nanoscale without excessive losses. The efficiency of the conventional near field aperture probe reduces with the 4th power of the aperture size; as a result the local intensity decreases much faster than the actual gain of confinement, i.e. one trades enhanced resolution for a very poor photon budget. Therefore the focus is on resonant metallic particles that enhance the local field enhancement and on nano-antenna configurations which might afford improved coupling efficiency: the field of nanoplasmonics. In parallel, new physics routes are explored through superlensing [6] by negative index (meta)materials, where the evanescent decay is locally inverted to gain; unfortunately, material losses are competing heavily with the superlensing efficiency.
Clearly, modern super-resolution imaging relies on creative exploitation of diffraction optics, where the role of efficient photo switchable fluorophores is crucial. Although sometimes the terms resolution and localisation are confused, it becomes clear that the finest detail one can discern will ultimately be determined by the amount of light, i.e. photon statistics.
[1] Method of the year 2008, Nature Methods 6, 1 (2009).
[2] E. Betzig, Proposed method for molecular optical imaging, Opt. Lett. 20, 237-239 (1995); E.Betzig et al., Imaging intracellular fluorescent proteins at nanometer resolution, Science 313, 1642-1645 (2006).
[3] M.J. Rust, M. Bates & X. Zhuang, Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM), Nature Methods 3, 793-796 (2006).
[4] S.W. Hell & J. Wichmann, Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy, Opt. Lett. 19, 780-782 (1994); V. Westphal & S.W. Hell, Nanoscale resolution in the focal plane of an optical microscope, Phys. Rev. Lett. 94, 143903 (2005); C. Eggeling et al., Direct observation of the nanoscale dynamics of membrane lipids in a living cell, Nature doi:10.1038/nature07596 (2008).
[5] M.V. Berry & S. Popescu, Evolution of quantum superoscillations and optical superresolution without evanescent waves, Phys. A: Math. Gen. 39, 6965-6977 (2006); F.M. Huang et al., Nanohole array as a lens, Nano Lett. 8, 2469-2472 (2008).
[6] J.B. Pendry, Negative refraction makes a perfect lens, Phys. Rev. Lett. 85, 3966-3969 (2000); N. Fang et al., Subdiffraction-limited optical imaging with a silver superlens, Science 308, 534-537 (2005).

THE VIEWPOINT by Niek van Hulst
Many Photons get More
out of Diffraction
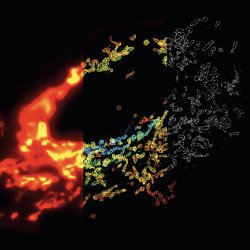
Nanoscopy, optical microscopy with 10-30 nm detail, has become a reality; in fact it has been chosen “Method of the year 2008” by Nature Methods [1]. We are all aware of the wave nature of light and the ensuing diffraction limit for resolution. So have the rules of physics been broken? What’s the trick and where’s the catch?STORM vs. Fluorescence Microscopy. Mitochondrial network in a mammalian cell visualized by 3D STORM. Conventional fluorescence image (left), 3D STORM image with colors denoting z location (middle) and single xy cross-section from the 3D STORM image (right). Copyright: Nature Methods 6, 17 (2009).
It is the capacity to detect and image single molecules at room temperature that has triggered ideas towards molecular optical imaging. Essentially, the position of a single isolated molecule can be determined with very high accuracy, limited only by the amount of detected signal. In fact, if N photons are detected, Poison statistics tells us that the location precision improves with N1/2, (assuming other noise terms can be ignored). For example, for 1000 counted photons the position accuracy is ~3% of the actual spatial response, which comes down to 15 nm for a diffraction limited spot of 500 nm. Already in 1994, Eric Betzig [2] had stated that if each molecule could be discriminated by some unique optical feature, a map of their positions could be reconstructed and molecular resolution imaging would become possible. It took another decade before a natural discriminator for molecules was exploited: time. Single molecules do blink and bleach; a stochastic process where only a fraction of the molecules is on at a time. To get control over the irregular time behaviour, Betzig and colleagues introduced photoactivation: with a delicate optical stimulus they activated a tiny fraction of molecules, imaged this low density fraction of molecules until they all photo-bleached and then they determined their exact positions. This way a flag is set for each molecule that dares to show up. By successive iteration of this activation-mapping-bleach trick they built a position map. The map gets filled with flags, all at nanometric accuracy through single molecule localization: Photo-Activation Localization Microscopy (PALM) [2]. An alternative approach is to switch molecules on temporarily, without bleaching, a method known as Stochastic Optical Reconstruction Microscopy (STORM) [3]. In either case, photo-switching microscopy is finding rapid spreading of applications in a variety of biological domains. PALM and STORM are advertised as super-resolution techniques, revolutionizing microscopy by breaking the diffraction limit for microscopy. However, isnt this revolution a deconvolution? No, it is not a simple deconvolution with the microscopic spatial response function, as the time discrimination of single photon emitters is essential: truly photoswitching microscopy. Has the diffraction limit been broken? Definitely not; nevertheless one should clearly differentiate between diffraction limited microscopic resolution and photon yield limited localization accuracy. It is the combination of high resolution and high signal that yields super localisation: all according to the rules of diffraction and photon statistics. Thus, the result of localization microscopy is a detailed map of discrete molecular positions, a reconstructed image, built-up through a stochastic process. The catch is that one relies on photoswitchable fluorescent molecules, detected at the level of single molecules, which are by necessity bleached for localization mapping.
STED vs. Confocal Microscopy. Comparison of confocal (Conf) and STED image of immunolabeled vimentin in a mammalian cell, after linear deconvolution. Nature Methods 6, 27 (2009).
Still, there is a soft edge to the diffraction limit. Recently, Michael Berry [5] showed that sub-wavelength optical fields can actually be created in the far field, at distances of many wavelengths. These so-called super-oscillations are a subtle phenomenon associated with delicate correlations between Fourier components. In principle, sub-wavelength focal points can be created at longer distances (not to be confused with the Talbot effect) without evanescent waves, based on these super-oscillations, containing spatial frequencies beyond the free space wave vector, i.e. faster than Fourier. Unfortunately the super-oscillations form a rapidly decaying tail of the total spatial frequency spectrum. Thus the high resolution components are exponentially weak in the super-oscillatory region and are surrounded by strong, low frequency components. If it wasnt for very good signal/noise allowing a high dynamic, one could not hope to discern some of the super-resolution aspects.
To break the diffraction limit, free of photodynamic properties of the sample, one needs to interact predominantly with the non-propagating, high spatial frequency contribution and by necessity move into the near field. Here, ironically, the sample becomes part of the microscope: one measures the spatial response of the mutual interaction and any super-resolution response cannot be specified as resolution of the microscope. For example, scanning a metallic nanoparticle around a luminescent quantum dot gives very informative nanoscale maps, but where is the microscope? A predominant challenge that remains in near field imaging is to confine the optical field to the nanoscale without excessive losses. The efficiency of the conventional near field aperture probe reduces with the 4th power of the aperture size; as a result the local intensity decreases much faster than the actual gain of confinement, i.e. one trades enhanced resolution for a very poor photon budget. Therefore the focus is on resonant metallic particles that enhance the local field enhancement and on nano-antenna configurations which might afford improved coupling efficiency: the field of nanoplasmonics. In parallel, new physics routes are explored through superlensing [6] by negative index (meta)materials, where the evanescent decay is locally inverted to gain; unfortunately, material losses are competing heavily with the superlensing efficiency.
Clearly, modern super-resolution imaging relies on creative exploitation of diffraction optics, where the role of efficient photo switchable fluorophores is crucial. Although sometimes the terms resolution and localisation are confused, it becomes clear that the finest detail one can discern will ultimately be determined by the amount of light, i.e. photon statistics.
[1] Method of the year 2008, Nature Methods 6, 1 (2009).
[2] E. Betzig, Proposed method for molecular optical imaging, Opt. Lett. 20, 237-239 (1995); E.Betzig et al., Imaging intracellular fluorescent proteins at nanometer resolution, Science 313, 1642-1645 (2006).
[3] M.J. Rust, M. Bates & X. Zhuang, Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM), Nature Methods 3, 793-796 (2006).
[4] S.W. Hell & J. Wichmann, Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy, Opt. Lett. 19, 780-782 (1994); V. Westphal & S.W. Hell, Nanoscale resolution in the focal plane of an optical microscope, Phys. Rev. Lett. 94, 143903 (2005); C. Eggeling et al., Direct observation of the nanoscale dynamics of membrane lipids in a living cell, Nature doi:10.1038/nature07596 (2008).
[5] M.V. Berry & S. Popescu, Evolution of quantum superoscillations and optical superresolution without evanescent waves, Phys. A: Math. Gen. 39, 6965-6977 (2006); F.M. Huang et al., Nanohole array as a lens, Nano Lett. 8, 2469-2472 (2008).
[6] J.B. Pendry, Negative refraction makes a perfect lens, Phys. Rev. Lett. 85, 3966-3969 (2000); N. Fang et al., Subdiffraction-limited optical imaging with a silver superlens, Science 308, 534-537 (2005).
Niek van Hulst
2009 © Optics & Photonics Focus
Niek van Hulst is ICREA Research Professor and senior group leader "Molecular NanoPhotonics" at ICFO - The Institute of Photonic Sciences in Barcelona (Spain).